写这个是因为我有个计数器,当同一个问题被问到一定次数后,我就会直接集中回答。
问:反应组学的一般分析流程是什么?
答:这个我在2021年匹斯堡会议上做过一个关于一般分析流程的口头报告,报告幻灯片在这里General reactomics data analysis framework for retrieving chemical relationship from untargeted mass-spectrometry data (yufree.github.io)。
基本有三步,第一步是基于质量差与相关性去除掉原始峰列表的冗余峰,如果你的峰表本来就不超过一万个峰,可以不做,这步主要是减轻计算负担。第二步是提取高频质量差,当然我感觉很多人用的时候其实是给出了自己关心的质量差,这步的目的是截取出样品化合物里潜在发生过的化学反应,同时可以通过定量pmd的方式去评估某类反应在不同分组样品里的强弱。第三步是通过将质量差连接起来形成网络来评估整体代谢过程,这一步的评估手段是利用网络拓扑参数,例如节点间平均距离等,这些参数代表的是某组样品整体反应的情况,也可以在进行物质鉴定的前提下用来讨论具体物质的代谢通路。
这个整体的分析流程目前可以在rmwf包里找到数据分析模版,里面有详细代码。
remotes::install_github('yufree/rmwf')
rmarkdown::draft("reactomics.Rmd", template = "reactomics", package = "rmwf")最近我注意到很多利用质量差进行的研究工作,这里建议不论你用不用pmd或是否引用我的工作,请使用reactomics或反应组来描述此类分析,使其区别于以物质鉴定为中心的代谢组学,不然你的新意很难被同行看到,也很难获得更大影响力。目前我的研究体会是很多学科都需要这个方法,但他们大都不懂质谱,使用反应组这个提法在交流上会有很大便利。
问:质谱反应组学跟质量亏损还有KMD有什么区别?
答:本质上没区别。质量亏损是发生在原子层面上的,当质子与中子结合形成原子时会损失掉一点质量,这就是爱因斯坦E=mc2为数不多能用上场的地方。形成质量亏损的核反应跟通常意义上的化学反应区别还是很大的,宇宙里所有元素的形成是经历过核反应的,本身也都有核结合能。单核结合能最高的是铁元素,因此所有元素都不如铁元素稳定,原子序数比铁高的会发生核裂变形成低原子序数元素,这就是当前核电厂的运行原理,当然原子序数比铁低的就要通过聚合形成序数高的元素,这就是核聚变,也是太阳发光发热的运行原理。这也是为啥有人说黑洞里面是个大铁块的原因,在黑洞的引力作用下所有元素都要形成最稳定的铁元素,不过还没形成黑洞前的中子星状态已经不存在原子的概念了,黑洞里究竟是啥,可能是数学吧。
原子层面的质量亏损是会累计到分子层面的,这样特定官能团的质量数就会非常独特,用高分辨质谱去测量就有较大把握给出元素组成。KMD图是一种基于质量亏损的可视化分析方法,简单说就是物质原有质量去乘一个归一化的质量差,例如最长用的质量差是CH2,归一化就是14/14.016,这样得到的Kendrick质量数会有一个特征,那就是遇到另一个跟其精确质量只差CH2的物质时,其Kendrick质量之差一定是这个归一化数的整数倍。这样你用Kendrick质量数做横轴,Kendrick质量之差做纵轴,如果两个物质在一条横线上,其质量差就是固定的。KMD这种图一般在FTICR这种贵族仪器上用的多,而能用上这个的一般是石化行业及相关机构拿来分析石油烃成分的,所以很长时间以来KMD都是用CH2做单元的,但不代表只能用CH2。
从反应组学角度,KMD图所做的就是将数据中所有质量差固定的物质提取出来,因此使用CH2的KMD图可看作反应组学的特例与一种可视化方法,而反应组学可看作更一般的质量亏损分析。例如,我现在想找出先进行羟基化,之后又进行甲基化的多级代谢物,就可以直接寻找所有质量差为15.995与14.016的质量对,如果质量对之间存在质量递增的关系,那么我们就事实上找到了这个多级代谢过程。KMD的方法只能找同系物,无法找不同质量差之间的关系,当然存在高阶质量亏损可视化分析,但反应组学理论上可以递归找到所有反应关系,而高阶质量亏损找三阶以上的计算上已经很麻烦了。如果你看不懂上面这句没关系,直接理解反应组学就够了。
问:反应组学是否可以用在纯高分辨质谱数据上?
答:可以。虽然我在开发pmd包的时候演示数据用的是色谱-质谱联用的峰表,但是也设置了 getrmd这个函数来处理纯质谱数据,其输入仅需要一个荷质比数值向量或者一行分子式,输出为一个行名为质量数、列名为质量差的真值表。如果你对某个质量差感兴趣,可以用真值表来过滤荷质比数值向量得到相关的荷质比。我刚更新了一个功能,如果你设置 verbose = TRUE,那么返回的就是一个列表对象,里面有两个元素,第一个是质量差表:第一列是高荷质比,第二列是低荷质比,第三列是质量差,这个表可用来找具体产生质量差的质量对,也可以用来生成质量差代谢网;第二个还是真值表。另外,这里的质量差是基于数据自身的高频质量差,你需要给出高频的定义,默认是10且最多选前20的质量差,我刚更新了一个功能,可以自定义一个质量差向量来进行筛选。
# remotes::install_github('yufree/pmd')
library(pmd)
data(spmeinvivo)
# 给定质量差且输出详细结果
re <- getrda(spmeinvivo$mz,pmd=c(2.016,15.119,18.011,14.016),verbose = T)## 4 pmd used.head(re$mdt)## ms1 ms2 diff diff2 md
## 19 118.1 100.1 18.011 18.011 0.01107
## 20 118.1 100.1 18.011 18.011 0.01101
## 19805 132.1 114.1 18.011 18.011 0.01088
## 24009 120.1 118.1 2.016 2.016 0.01601
## 25411 120.1 118.1 2.016 2.016 0.01595
## 46359 153.1 135.1 18.011 18.011 0.01091head(re$result)## 2.016 14.016 15.119 18.011
## 100.076307947972 FALSE FALSE FALSE TRUE
## 100.510657997265 FALSE FALSE FALSE FALSE
## 100.957164176016 FALSE FALSE FALSE FALSE
## 103.054737631236 FALSE FALSE FALSE FALSE
## 104.009041714793 FALSE FALSE FALSE FALSE
## 104.108169555664 FALSE FALSE FALSE FALSE有这个疑问的一般是进行溶解性有机质研究的,而且也用的是FTICR这种仪器,给出的结果连分子式都有。如果你读过我的原始论文应该知道我计算质量差数据库其实就是先算了分子式差异,然后累加的差异原子最高丰度的同位素精确质量。所以这里算质量差其实有点大材小用,我建议直接算分子式差异,这样后面解释反应更方便。具体代码在我原始论文里有,当然重写也不困难。
不过,这里的问题在于没有了色谱分离,如何排除干扰性质量差的影响。这里所谓干扰性质量差,就是随便找两个荷质比做差值形成的质量差,这种质量差可能并不具备反应属性,单纯就是算差值时算出来的。这种情况对色谱质谱联用的数据影响不那么大,那边可以通过色谱分离区分来自物质自身的质量差与不同物质间的质量差,但对于纯质谱数据,当两者混到一起在计算质量差时会出现很大的冗余问题,
这里我依然采用质量亏损原理来过滤,具体说就是看质量差小数点后位小数是否小于0.25或大于0.9,如果处于这一区间,那么这个质量差大概率是真实物质间质量差。我计算过12329个已知反应的质量差,只有不到5%的质量差在这个范围之外,下图是已知反应质量差小数点后小数的分布。
data("omics")
pmd <- omics$pmd[!is.na(omics$pmd)]
# 计算质量差的质量亏损
pmdmd <- pmd%%1
# 计算比例
sum(pmdmd>0.25&pmdmd<0.9)/length(pmdmd)## [1] 0.04769# 展示分布
hist(pmdmd)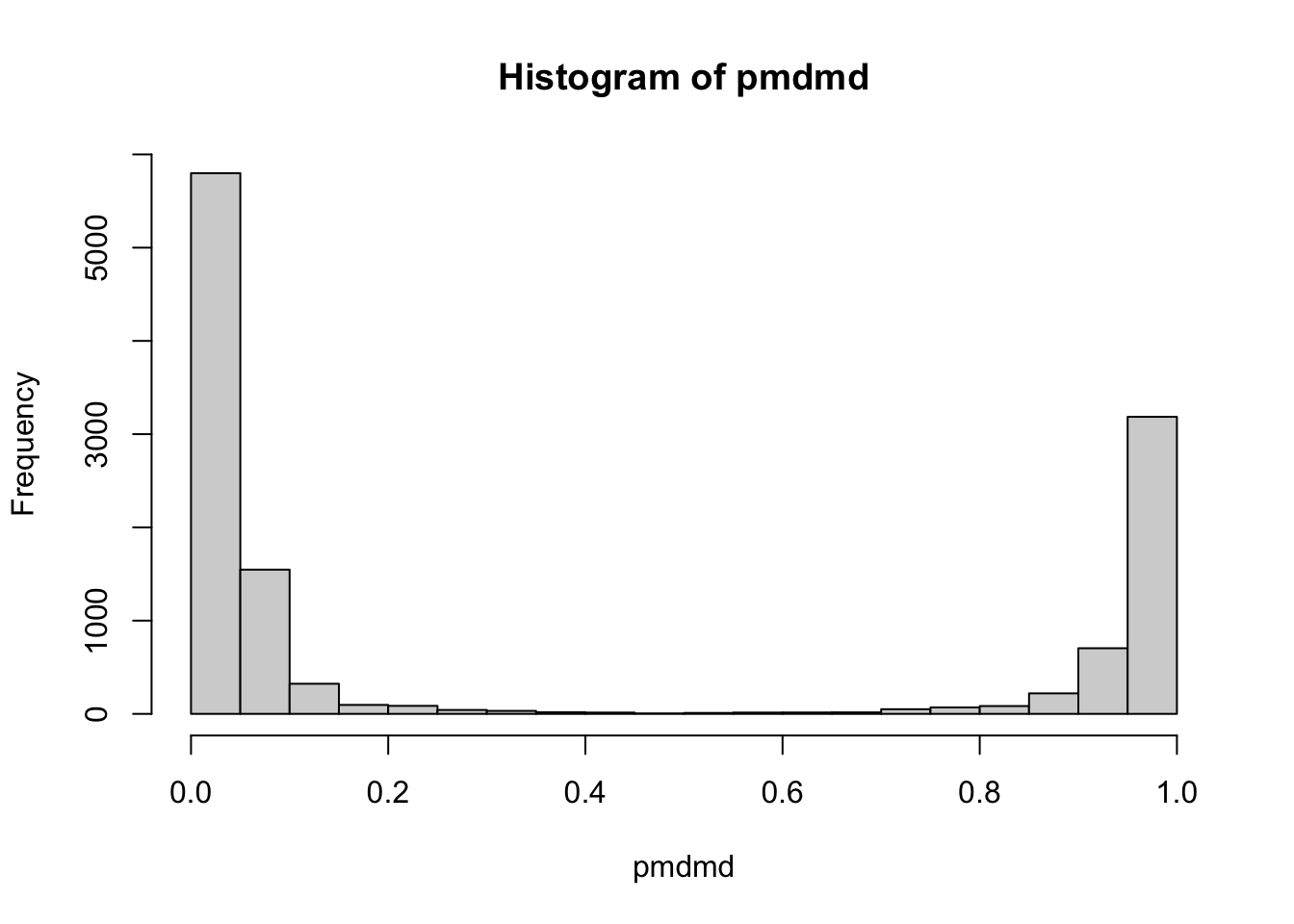
这个范围作为筛选问题不大,而且在使用函数时你可以改掉这个范围或者设置为NULL不进行筛选。
至于说同位素的影响,这块我没有在函数中体现,不过函数返回的表是你去找质量差为1.002的那一列应该就是可能的同位素峰了。我没有设置同位素去除的功能主要是考虑到测量上的问题,即使FTICR的数据,同位素质量差也会在0.001到0.003之间波动,主要原因在于质量差测量会累积两个质量数测量上的不确定度。我在这里不做处理主要是让研究人员根据自己仪器情况自由发挥。不过这个过滤后结果已经足够进行后续分析了。
问:我按照 pmd 包中的 getrda 函数计算出了分子之间相对应的质量差并在pmd 包提供的kegg库中利用比对到了,但是对于同一个质量差我有很多化合物而kegg库中只有一个转化反应,还跟我的分子式不一样,对于我没有比对出来的分子之间的转化可以理解为只要pmd值相同则他们反应所需的酶或者蛋白是一样的吗?
答:kegg只是已知反应的库,包里面带这个主要也是为了找一下已知的质量差,至于说具体母体化合物与产物能不能对上其实不太重要。可以解释为酶或蛋白类似,不放心可以去验证,不过如果都去验证那意义也不大了,环境样品里大多数物质是没法验证的,主要看他们整体变化趋势是否随你实验设计有变化。
问:对于一个化合物,其代谢为不同化合物会对应不同的pmd值,根据这个化合物和相应的pmd值我可以通过构建pmd连接的峰网络去计算链接网络的平均距离,如果平均距离越长,就代表这个物质来自内源生化反应吗?另外这个距离的长短是如何定义的,还是说他只是一个相对大小比如A比B的平均距离更长,则A比B更可能来源于内源生化反应?
答:这个平均距离是针对具体样品关心的化合物与质量差而谈的。给定具体高频反应或质量差例如2.02,15.99,18.01,14.02,如果某个物质形成的pmd网络节点间平均距离越长,越说明样品本身存在对应的多步代谢过程,而生物体内进行多步代谢的小分子一般是内源化合物。如果是外源污染物,其对体内代谢过程是相对陌生的,因此代谢一两步可能也就排出体外了。这里不是绝对的,只是从概率上更可能,但具体到平均网络长度,高频质量差下超过5大概率就是内源化合物了。这里需要注意,同一个物质在不同生物样本里角色可以不一样,有些物质可以被生物A代谢但不被B代谢,那么对于生物A就算内源化合物,B算外源化合物。
问:如果我有一个化合物,想看其在某一组质量差下的代谢物,我可以直接构建pmd网络,然后探究可能潜在代谢物,是这样吗?
答:是的。pmd包里是通过getchain这个函数来实现的,给定峰列表、母体化合物荷质比与质量差,函数会以母体化合物荷质比为起点搜索所有符合质量差的峰,然后再以找到的峰为起点继续寻找符合质量差的峰直到无法找到新的峰后程序终止。这样你会得到一个含有质量差起点与终点的数据框,基于此可以构建代谢网络并可视化。
问:筛选高频pmd值构建网络的目的是什么呢?
答:筛选高频pmd主要考虑的是样品自身的特性,反复出现的pmd说明样品中存在对应的酶,这种筛选主要是为了探索样品自身独特的那一组反应集合。当然,实际样品里常见的高频pmd其实大多跟kegg库里的高频pmd是重合的,这体现了生化反应类型的保守性,所以你去指定一组pmd也不会有问题。如果你发现某个pmd频率特别高,但其实不常见,那说明这个样品里含有对特定反应敏感的酶或代谢通路,如果这些物质在不同样品里变化趋势跟你的实验设计存在对应关系,那么说明你的实验处理可能激活或抑制了某种反应,后面就要具体情况具体分析了。目前我给的定量方法就是把相同质量差的分子且响应比变化不大的直接加合起来,这样一个质量差会有一个综合响应,如果这个综合响应在实验组对照组出现差异,那么可认为这个反应被激活或抑制了,进而可以讨论些代谢通路的影响了。
大家如果有其他关于反应组学的疑问,可留言或邮件咨询。来问过我的老师同学应该都清楚,我不会去要求什么冠名权,也不会藏着掖着,很多提出的问题我都想过很多遍了。很多软件里函数的功能其实远超出最初发表的文章内容但我没空去整理发表,只有问多了我才会觉得重要然后集中回答一波。
ps. 上面用到的pmd包不是CRAN上的,而是我GitHub上的开发版。