原始数据导入时我们所遇到的背景问题说到底是测定的系统误差,期望可以看作0。但有些影响是我们不希望看到但依旧存在的,例如你测定了100多份基于地区分布的人体样本中的感兴趣基因组,但由于采样原因不可能同时采集,而时间的差异会直接导致诸如温度等影响了样本的均质性,这种情况下进行统计推断就需要平衡掉这些因素的影响。那么,如何屏蔽呢?
首先我们要推广下t检验到线性回归。两组数据的差异比较可以构建如下模型:
$$Y = \alpha * X + \beta$$
Y是响应,假设有两组,我们给组A赋值1,组B赋值0,那么系数$\beta$就是组B的均值而系数$\alpha$则是组A与组B的差值。这个差值实际上就是t检验里的差异,对这个系数的估计出的t值就是t检验的t值。
下面运行下代码看看
# 模拟生成两组数据
set.seed(42)
group1 <- rnorm(100,100,10)
group2 <- rnorm(100,130,13)
# 进行t检验
t.test(group1,group2)
##
## Welch Two Sample t-test
##
## data: group1 and group2
## t = -18.173, df = 195.17, p-value < 2.2e-16
## alternative hypothesis: true difference in means is not equal to 0
## 95 percent confidence interval:
## -31.63463 -25.44050
## sample estimates:
## mean of x mean of y
## 100.3251 128.8627
# 构建分组变量
fac <- c(rep(0,100),rep(1,100))
# 构建数据变量
dat <- c(group1,group2)
# 回归分析
fit <- lm(dat~fac)
summary(fit)
##
## Call:
## lm(formula = dat ~ fac)
##
## Residuals:
## Min 1Q Median 3Q Max
## -30.256 -6.523 0.566 6.532 36.262
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) 100.33 1.11 90.35 <2e-16 ***
## fac 28.54 1.57 18.17 <2e-16 ***
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 11.1 on 198 degrees of freedom
## Multiple R-squared: 0.6252, Adjusted R-squared: 0.6233
## F-statistic: 330.2 on 1 and 198 DF, p-value: < 2.2e-16
上面我们可以看到fac的参数估计得到的t值与t检验是一样的,截距也就是组B的均值,而斜率则是组A与组B的差。由此我们可以看到t检验可以看作线性回归针对两组变量的一个特例。而进入线性回归领域我们可以做的东西就多了,例如我们平时所说的单因素方差分析与多重比较本质上就是线性回归针对多组变量的特例。而线性回归更多是用在处理连续变量的,那么本质上你的分组也可以是连续的。扯远了,在基因组相关分析上我们引入回归分析多半不是针对样本分组的,而是用来平衡影响因素的。
回到我们简单的两组差异比较上问题可以描述为两组样本的采集并不随机。在测定身高问题上,当我们采集样本时间超过1年,那么不同年份测定的身高会受身高自然生长(青少年)的影响。那么我们在采集完数据后不能因为采集时间过长而重新采集,而这在很多实验条件下也是不可能完成的任务。那么我们可以先探索性的分年份观察下数据,如果存在年份差异,那么我们可以构建下面的模型:
$$Y = \alpha * X + \gamma * Year + \beta$$
这样就相当于把采样时间纳入模型之中。数据处理时对年份数据我们进行归一化处理,这样均值也就为0。当我们解释$\beta$时,可以完全忽视年份的影响,而这样得到的$\beta$实际意义就是在一个平均年上的均值,由此年份产生的影响就消除了。本质上,这是一个通过将影响因素随机化的手段,都随机化掉也就更接近我们想知道问题的答案。同样的方法也常用于流行病学研究,例如考察影响心血管疾病发病率因素的模型都会把是否吸烟添加到生存分析的模型中,所以在文献报道中常出现的“考虑了吸烟,肥胖等因素后”实际上在数据层就是通过在回归分析里加入影响因素来去除影响的。
但是上面那是知道影响是什么,如果不知道呢?也就是不知道不知道的因素如何调整?完全不知道的话也就不调整了,但如果你知道存在但无法描述呢?最常见的就是背景干扰,我反正也不去描述,只要这个背景稳定我们直接扣除就完了,好比实验随机化了。眼不见当然心为净了,但我们其实可以分分钟给个热图出来,这时候我们就会发现,其实不能描述的影响还是很多的。
下面用人种分性别数据来观察下这个问题,我们已知的混杂变量是测定时间,分组变量是性别。
# 安装bioconductor
source("http://bioconductor.org/biocLite.R")
biocLite()
# 安装绘图用的rafalib包跟RcolorBrewer包还有示例的GSE5859Subset数据集
# install.packages('rafalib')
# install.packages('RColorBrewer')
# devtools::install_github("genomicsclass/GSE5859Subset")
# genefilter包用来同时进行高维数据操作
# biocLite('genefilter')
# 加载软件包与数据集
library(GSE5859Subset)
library(rafalib)
library(genefilter)
library(RColorBrewer)
# GSE5859Subset数据集收集了不同人种的基因表达数据的一部分
data(GSE5859Subset)
# 按照性别分组
sex <- sampleInfo$group
# 提取混杂变量时间
batch <- factor(format(sampleInfo$date,"%m"))
# 提取染色体编号
chr <- geneAnnotation$CHR
# 对不同时间进行t检验观察混杂是否存在
tt<-rowttests(geneExpression,batch)
# 提取Y染色体
ind1 <- which(chr=="chrY") ##real differences
# 提取时间尺度上不在Y染色体上的变化最大的50个基因中
ind2 <- setdiff(c(order(tt$dm)[1:25],order(-tt$dm)[1:25]),ind1)
# 从上面两组基因之外再随机选50个基因
set.seed(1)
ind0 <- setdiff(sample(seq(along=tt$dm),50),c(ind2,ind1))
# 这三组基因提取的各代表一定有变化的(Y染色体女性没有),采样时间导致的变化以及其他随机找的50个基因
geneindex<-c(ind2,ind0,ind1)
# 截取相应数据
mat<-geneExpression[geneindex,]
# 归一化
mat <- mat -rowMeans(mat)
# 绘图
mypar(1,2)
# 所有截取的数据
icolors <- colorRampPalette(rev(brewer.pal(11,"RdYlBu")))(100)
image(t(mat),xaxt="n",yaxt="n",col=icolors)
# 所有数据归一化
y <- geneExpression - rowMeans(geneExpression)
# 绘制样本间分性别相关趋势图
image(1:ncol(y),1:ncol(y),cor(y),col=icolors,zlim=c(-1,1),
xaxt="n",xlab="",yaxt="n",ylab="")
axis(2,1:ncol(y),sex,las=2)
axis(1,1:ncol(y),sex,las=2)
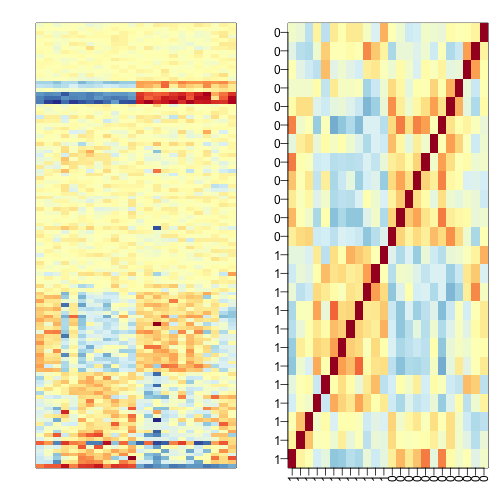
这张图上我们可以看出原始数据上面至少有两种模式可用来校准偏差,单纯考虑年份影响并不能解决所有的混杂模式,另外样品按性别去看相关也很难看出单一的趋势。那么如何把这些看得到但不好用分类或连续变量剔除的混杂因素给找出来呢?
这种情况要考虑使用主成分分析降维,当维度比较低的时候,单个维度都说明一种模式,数学上更有利的是主成分之间是正交的。但这样做有一个Bug,那就是你处理前是不知道哪个主成分会包含真实效果的,而扣除掉占主要方差的主成分后你会欣喜的发现剩下的全是噪音了。那么问题来了:一方面要保证主要效果不能丢失(但可能已经掩盖在噪音里了),另一方面还要把其余的混杂尽可能的删掉,有没有这样的分析方法呢?
有,替代变量分析。当我们不知道混杂变量是什么先用主成分分析对除了真实效果的那部分矩阵提取主成分,然后提取主成分在每一个基因上的投影作为权重,把权重迭代到原来矩阵上,然后在进行回归,直到转化的因子方差不怎么变化为止。这样实际上就是不断通过加权强化混杂变量的影响,最后在不清楚混杂变量模式的情况下把它们通过迭代找出来作为一个整体替代变量排除掉。类似的分析方法也有不少,不过我就理解了这一种,殊途同归,自行理解。
下面继续上面的案例,看看效果。
# 安装相关软件包sva与limma
# biocLite(c('sva','limma'))
# 读取进行替代变量分析与回归的sva跟limma包
library(limma)
library(sva)
# 构建基础模型
mod <- model.matrix(~sex)
# 寻找替代变量
svafit <- sva(geneExpression,mod)
## Number of significant surrogate variables is: 5
## Iteration (out of 5 ):1 2 3 4 5
# 构建包含替代变量的模型
svaX<-model.matrix(~sex+svafit$sv)
# 拟合模型
lmfit <- lmFit(geneExpression,svaX)
# 提取替代变量矩阵
Batch<- lmfit$coef[geneindex,3:7]%*%t(svaX[,3:7])
# 提取效应矩阵
Signal<-lmfit$coef[geneindex,1:2]%*%t(svaX[,1:2])
# 提取误差矩阵
error <- geneExpression[geneindex,]-Signal-Batch
# 归一化作图
Signal <-Signal-rowMeans(Signal)
mat <- geneExpression[geneindex,]-rowMeans(geneExpression[geneindex,])
mypar(1,4,mar = c(2.75, 4.5, 2.6, 1.1))
image(t(mat),col=icolors,zlim=c(-5,5),xaxt="n",yaxt="n")
image(t(Signal),col=icolors,zlim=c(-5,5),xaxt="n",yaxt="n")
image(t(Batch),col=icolors,zlim=c(-5,5),xaxt="n",yaxt="n")
image(t(error),col=icolors,zlim=c(-5,5),xaxt="n",yaxt="n")
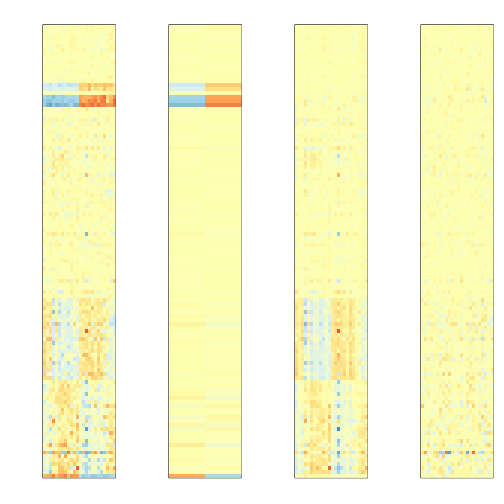
可以看出,通过替代变量分析我们最大程度上保留了原有效应并剔除了替代变量。在这个基础上配合前文提到的经验贝叶斯方法进行基因筛选,得到的基因为我们比较感兴趣的部分了,下一篇将在此基础上讨论如何对结果进行注释或者基因组序列比对。
在进行下一部分之前,我们有必要讨论下一些基因组数据分析的可视化方法,这些方法不见得新,但有利于我们发现数据本身的问题而不是蒙着头只凭过往经验做分析。
可视化
基因组数据的结构决定其可视化必然要适应高维属性并面向实际问题。下面分问题阐述且前面提到的火山图与热图就不单独介绍了:
重复性问题
你进行了一组技术重复,然后发现两组数据相关性不错,那么是不是数据就可信了?不一定,第一次你测到是1,3,5,7但第二次测到的是10,30,50,70。这种情况相关性非常好,但都差了一个数量级。我们用数据做个演示。
# biocLite("SpikeInSubset")
library(SpikeInSubset)
# 载入原始数据
data(mas95)
# 提取表达谱信息并用散点图绘制
mypar(1,2)
r <- exprs(mas95)[,1] ##original measures were not logged
g <- exprs(mas95)[,2]
plot(r,g,lwd=2,cex=0.2,pch=16,
xlab=expression(paste(E[1])),
ylab=expression(paste(E[2])),
main=paste0("corr=",signif(cor(r,g),3)))
abline(0,1,col=2,lwd=2)
# 寻找95%的数据集中的部分
f <- function(a,x,y,p=0.95) mean(x<=a & y<=a)-p
a95 <- uniroot(f,lower=2000,upper=20000,x=r,y=g)$root
abline(a95,-1,lwd=2,col=1)
text(8500,0,"95% of data below this line",col=1,cex=1.2,adj=c(0,0))
# 在对数范围上重新绘图
r <- log2(r)
g <- log2(g)
plot(r,g,lwd=2,cex=0.2,pch=16,
xlab=expression(paste(log[2], " ", E[1])),
ylab=expression(paste(log[2], " ", E[2])),
main=paste0("corr=",signif(cor(r,g),3)))
abline(0,1,col=2,lwd=2)
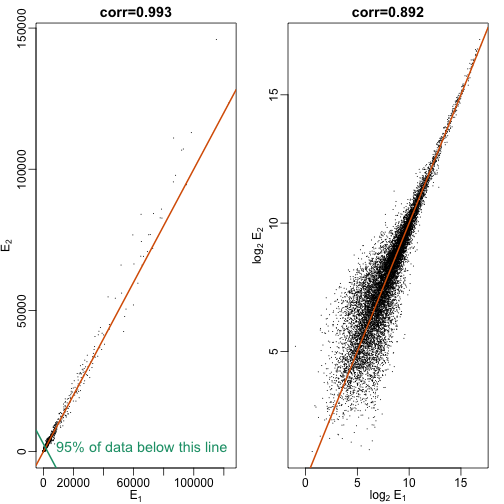
从上面的图中我们可以看出基因组技术重复数据大部分位于低响应区,且数值越小,方差越大。这种情况得到的相关性可能被少数数据所影响,因而我们采用绝对差值与均值做图给出比较。
mypar(1,1)
plot((r+g)/2,(r-g),lwd=2,cex=0.2,pch=16,
xlab=expression(paste("Ave{ ",log[2], " ", E[1],", ",log[2], " ", E[2]," }")),
ylab=expression(paste(log[2]," { ",E[1]," / ",E[2]," }")),
main=paste0("SD=",signif(sqrt(mean((r-g)^2)),3)))
abline(h=0,col=2,lwd=2)
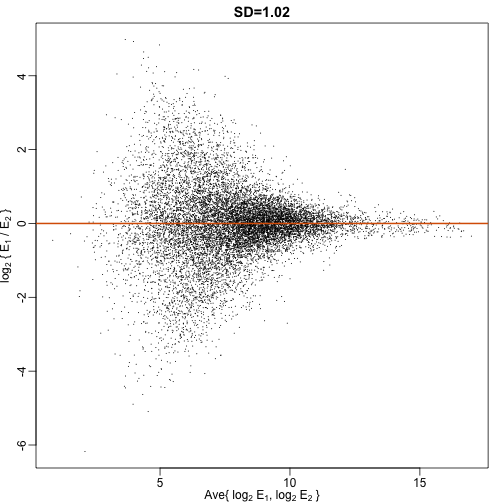
这样的图叫做MA-plot或者说Bland-Altman plot,在基因组数据分析文章中算是比较常见。从图上我们知道,两者对数差的平均方差为1,也就是说差距约为2倍。这个2倍所说明的重复性问题需要按实际情况来看。
异常值
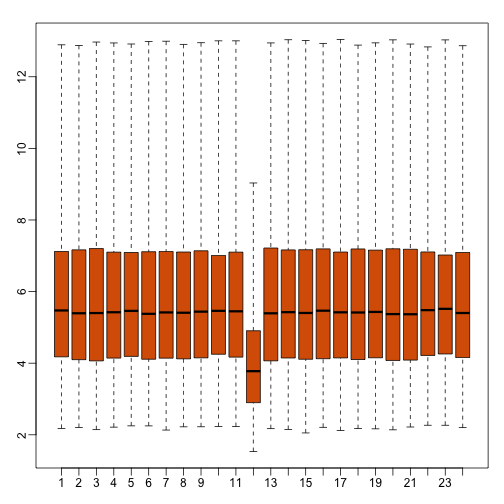
拿到数据后可以先来个最直观的boxplot异常值检测,出现特别离谱的,多半是有大问题。
ge <- geneExpression
# 伪造一组有问题的数据
ge[,12] <- ge[,12]/log2(exp(1))
mypar(1,1)
# 用boxplot检查
boxplot(ge,range=0,names=1:ncol(ge),col=ifelse(1:ncol(ge)==42,1,2))
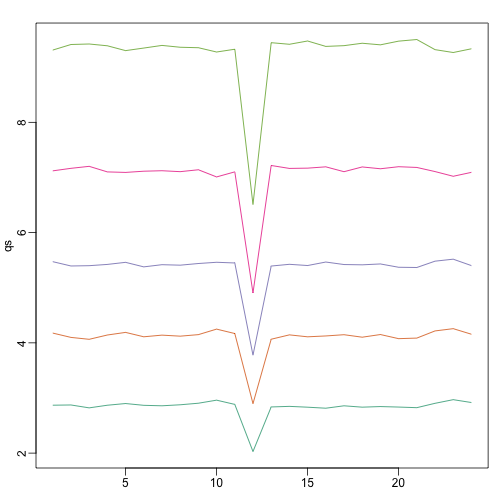
# 用matplot绘制分位数曲线
qs <- t(apply(ge,2,quantile,prob=c(0.05,0.25,0.5,0.75,0.95)))
matplot(qs,type="l",lty=1)
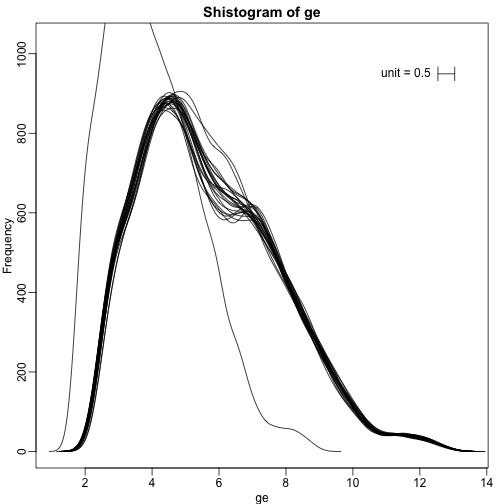
# 绘制平滑直方图
shist(ge,unit=0.5)
高维数据展示
除了热图以外,其实也有其他展示高维数据的方法,基本都是基于主成分分析。为了展示方便,一般都是截取前两个主成分,然后用颜色标记出样本类别,在R中可以用cmdscale方便的给出两个主成分。这是一种综合展示方法,纯描述探索性的,用来发现问题。
另外一种展示是基于聚类分析的,也就是当我不知道分类时,我可以用聚类先聚出几类来,然后画出来看看样子。当然也可以用来寻找变化相近的基因与某种器官或功能的联系。总之一图胜千言,看到了问题才好解决。